Log in To see your account pricing.
EpiTect Hi-C Kit
For high-resolution mapping of chromatin folding, high-quality assembly of genome sequences, haplotype phasing and identifying chromosomal rearrangements
For high-resolution mapping of chromatin folding, high-quality assembly of genome sequences, haplotype phasing and identifying chromosomal rearrangements
✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
Cat. No. / ID: 59971
✓ 24/7 automatic processing of online orders
✓ Knowledgeable and professional Product & Technical Support
✓ Fast and reliable (re)-ordering
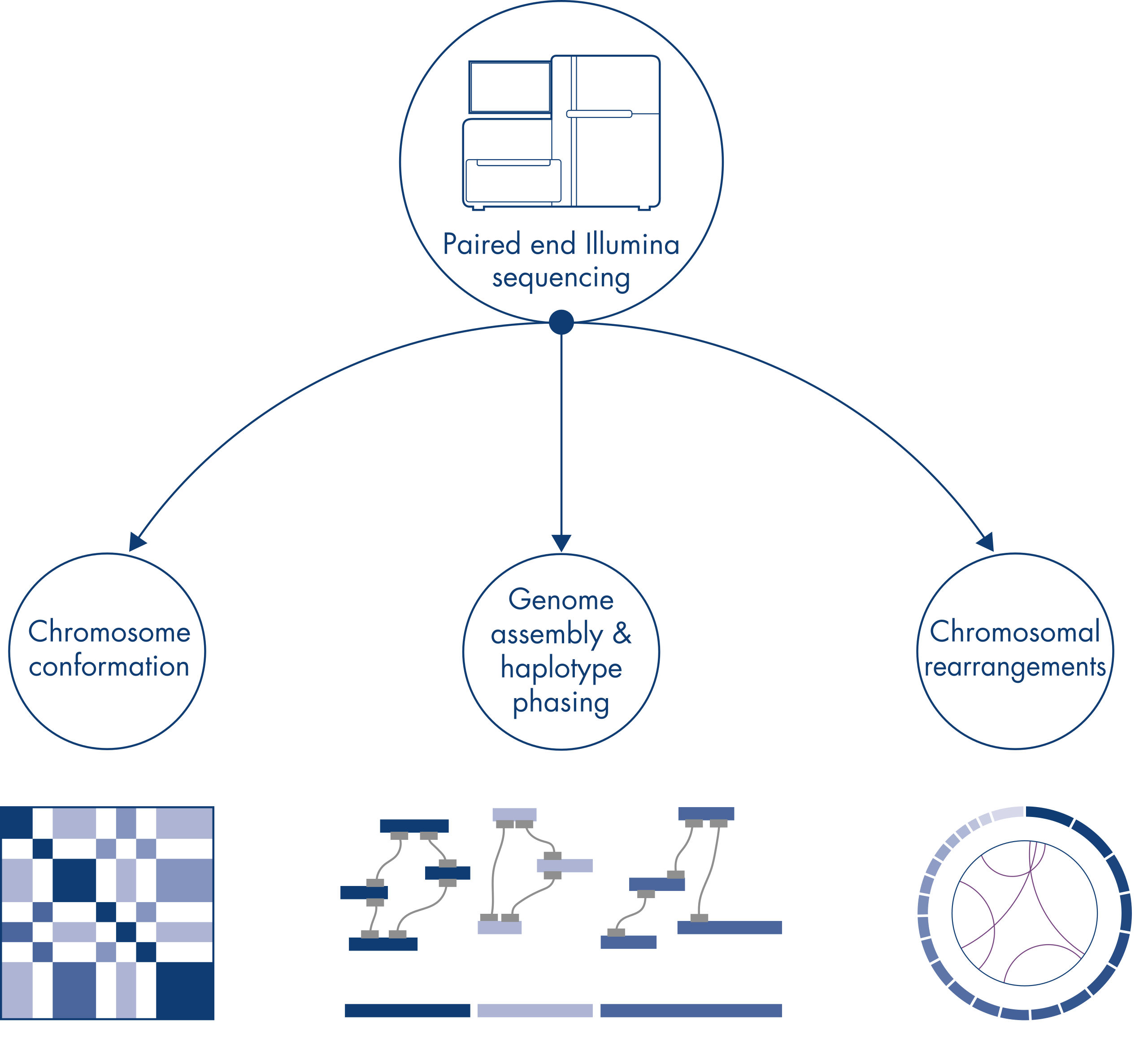
Hi-C was originally conceived as a powerful technique for genome-wide chromosome conformation capture, enabling the characterization of chromatin folding at kb resolution. However, the technology also has other important applications. For example, Hi-C is used for generating highly contiguous genome assemblies, with few and very long scaffolds, from organisms without a known reference genome. In addition, Hi-C is also very useful for haplotype phasing and detection of chromosomal rearrangements.
The EpiTect Hi-C Kit offers a robust, yet simple and fast, protocol with low cell input requirements that enables generation of high-quality Hi-C Illumina NGS libraries from cross-linked cells in less than 2 days.
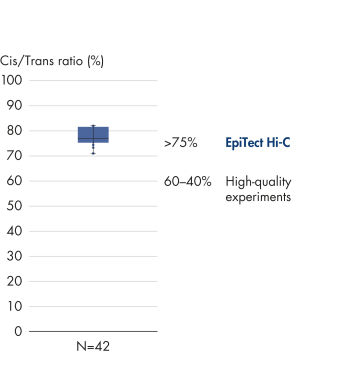
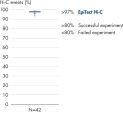
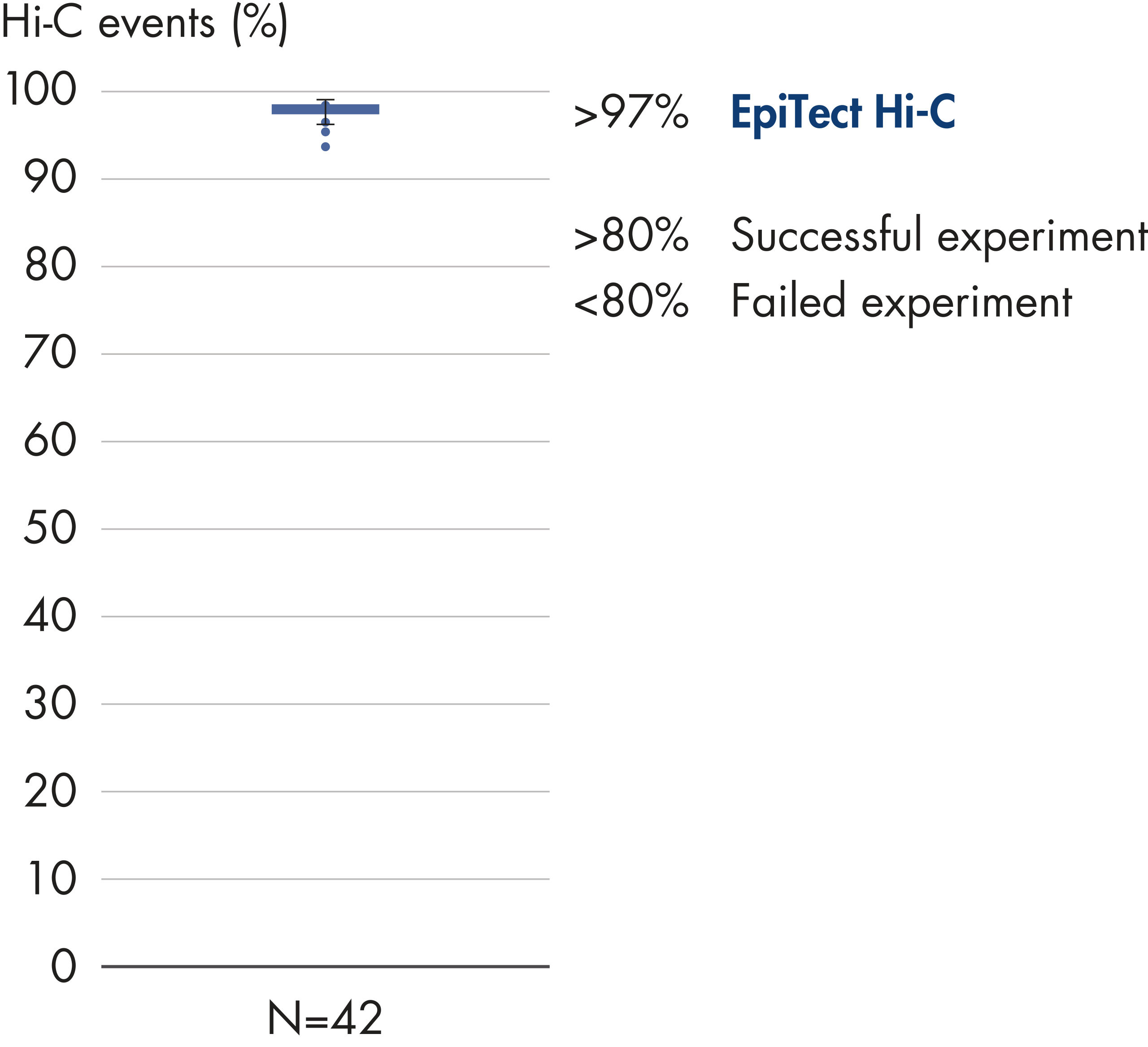
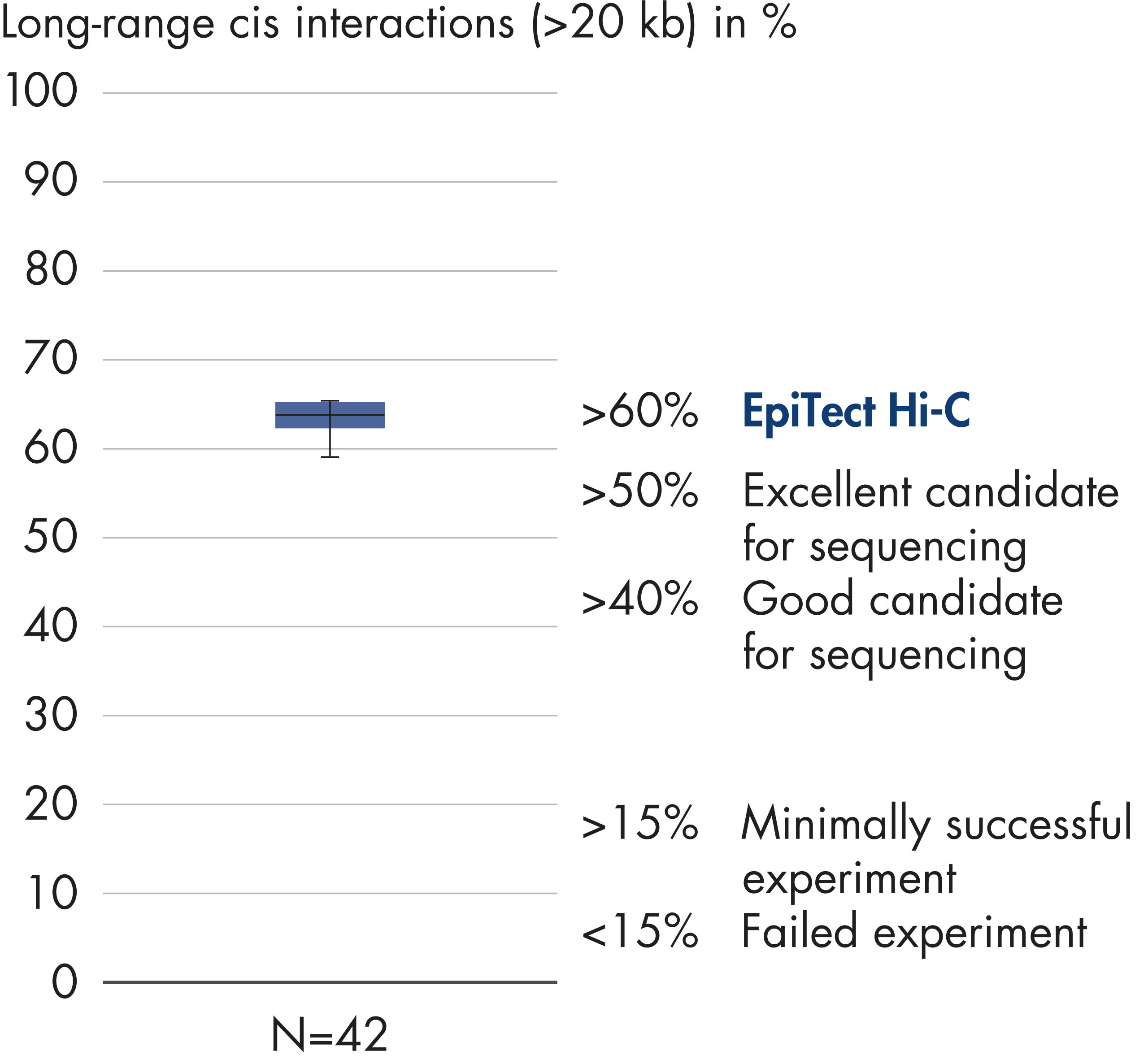
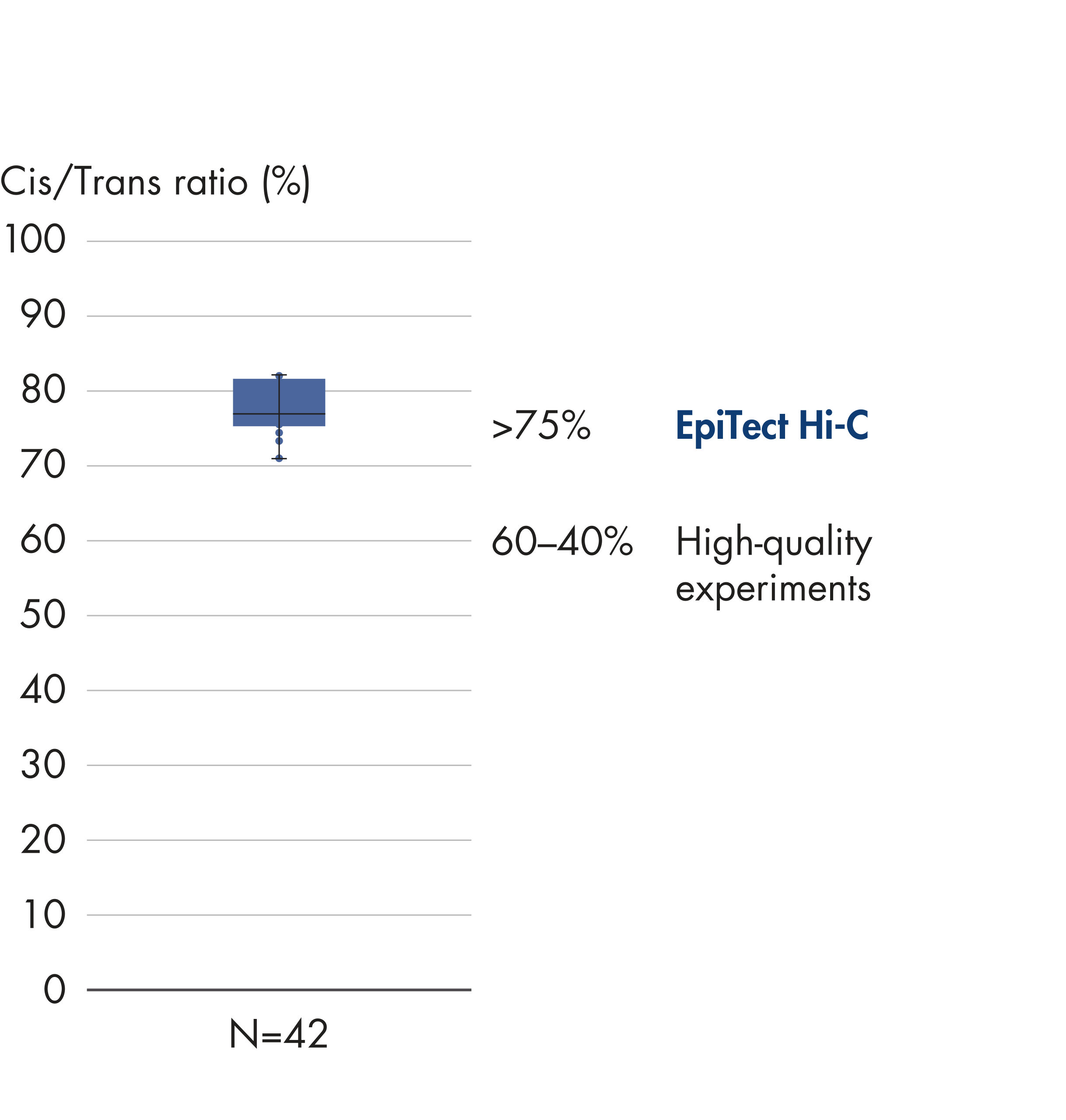
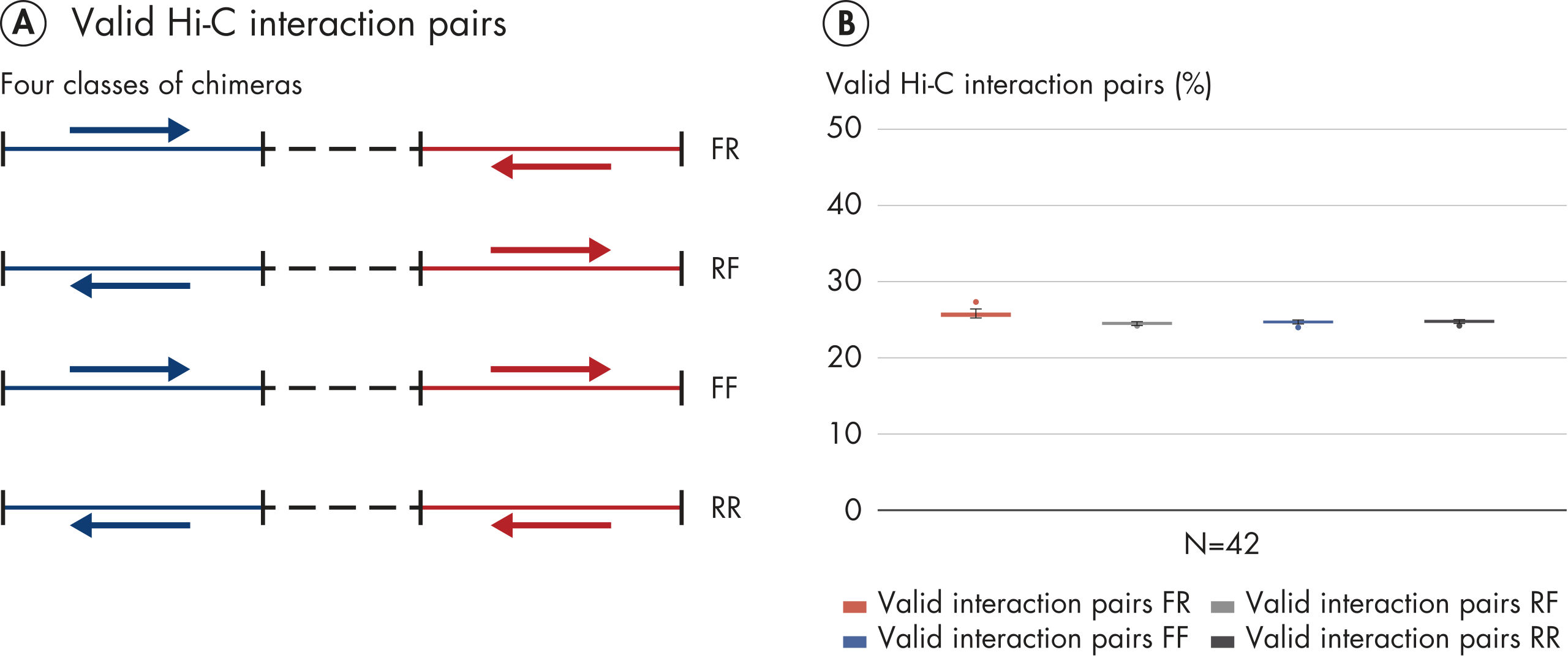
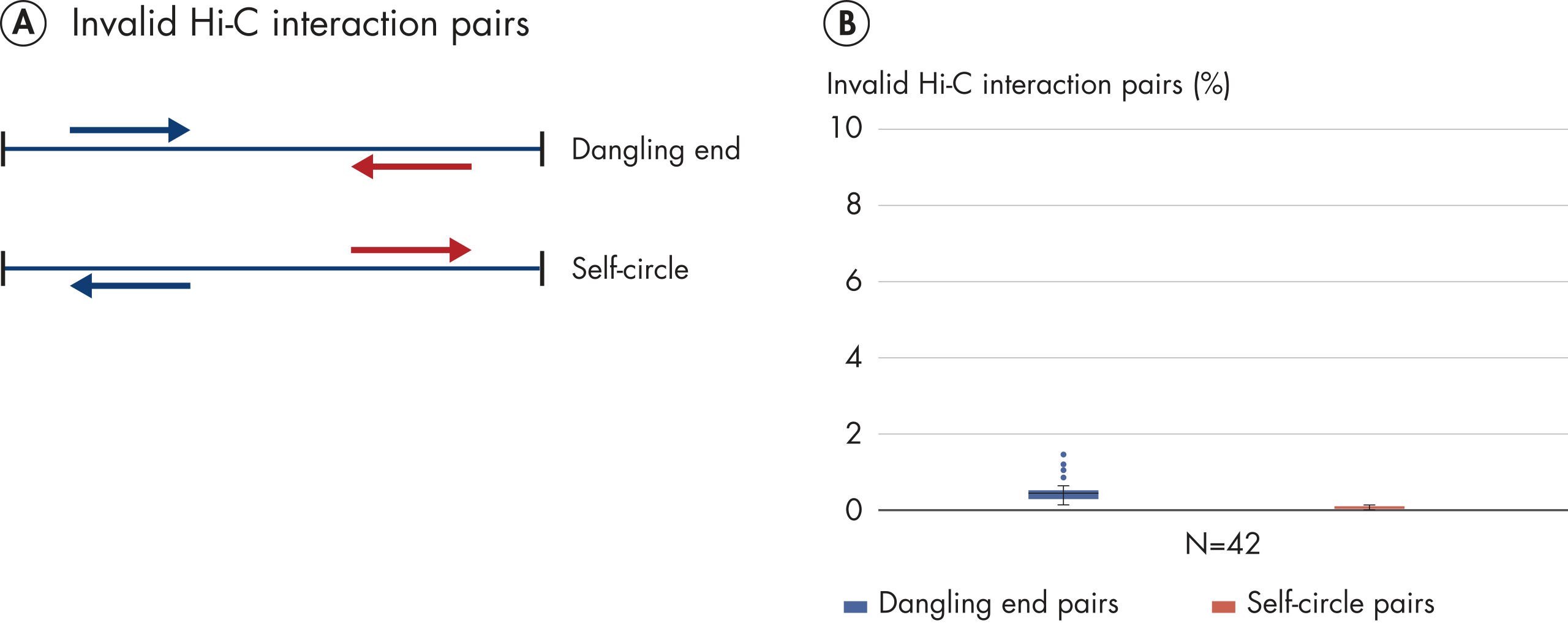
The EpiTect Hi-C Kit generates high-quality Hi-C NGS libraries, ensuring that first-rate data is generated from costly downstream deep sequencing. Sequencing results from more than 40 EpiTect Hi-C libraries have been analyzed to evaluate the performance of the kit. The most important QC metrics are shown in the following figures: Percentage of Hi-C events, Percentage of long-range cis interactions, Cis/Trans ratio, No strand orientation bias with the EpiTect Hi-C Kit and Percentage of paired reads deriving from a single restriction fragment. The data show that the EpiTect Hi-C Kit generates NGS libraries that, on average, far exceed criteria normally considered sufficient for a successful Hi-C experiment.
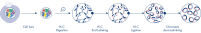
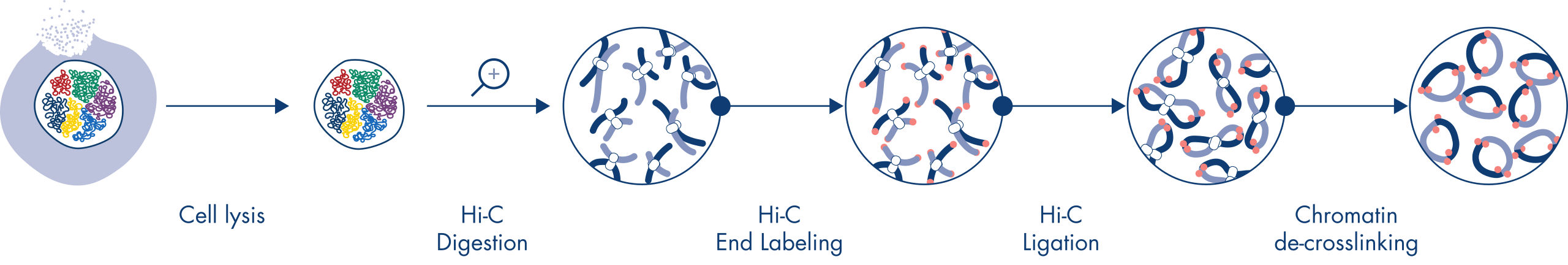
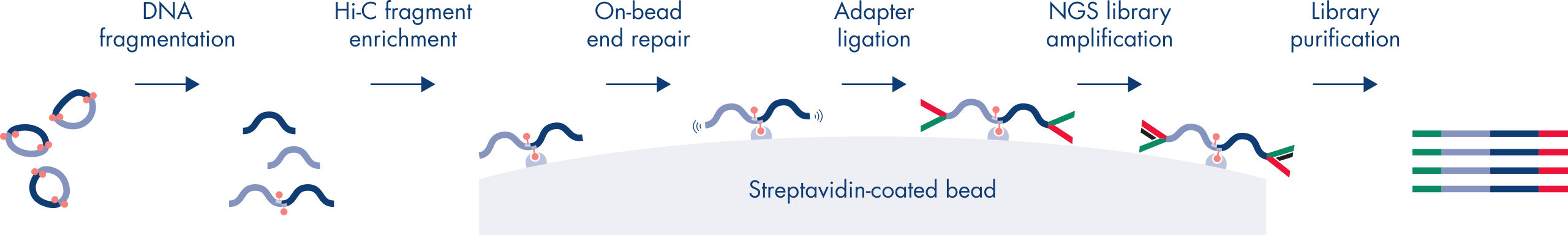
Hi-C is a proximity ligation assay that captures chromatin interactions on a genome-wide scale. The EpiTect Hi-C Kit is a specialized DNA preparation resulting in an Illumina-compatible NGS library (see figures EpiTect Hi-C workflow – day 1 and EpiTect Hi-C workflow – day 2). Briefly described, the assay starts with the purification of nuclei in which chromatin conformation has been frozen by chemical cross-linking of DNA binding proteins and DNA. The DNA is then completely digested with a 4 bp restriction enzyme. Open DNA ends are labeled with biotin and subsequently ligated. Paired-end sequencing of the Hi-C ligation products identifies very large numbers of chimeric sequences that derive from DNA strands that were closely associated in space. The probability that two sequences are ligated together is a function of their average distance in space. Quantification of ligation junctions allows for the determination of DNA contact frequencies from which high-resolution mapping of chromatin folding can be achieved.
The EpiTect Hi-C workflow (see figures EpiTect Hi-C workflow – day 1 and EpiTect Hi-C workflow – day 2) represents a marked improvement over published protocols. A week-long and complicated procedure has been converted into a simple and robust protocol that requires just 1.5 days. Furthermore, the sample input requirement has been reduced by one order of magnitude, allowing creation of Hi-C NGS libraries from just 5000 cells. The protocol has been developed for work with cross-linked cells from mammalian cell cultures.
The EpiTect Hi-C procedure is a version of the in situ (i.e., in nucleus) Hi-C method in which nuclei are gently purified and permeabilized to maintain the spatial organization of the genome during the initial digestion and ligation steps. This process is vital in order to minimize background noise from uninformative ligation events that do not reflect genome organization. This is because intact nuclei constrain the movement and random collisions of cross-linked complexes, such that ligation events predominantly occur between topologically associated DNA fragments.
Constructing Hi-C NGS libraries
The EpiTect Hi-C Kit workflow consists of 2 parts and each can be completed in one day. The steps of the protocol are summarized in the tables below and visualized in figures EpiTect Hi-C workflow – day 1 and EpiTect Hi-C workflow – day 2. The included Illumina adapters have sequence bar codes that enable multiplex sequencing of up to 6 samples.
To view the full protocol, see our detailed EpiTect Hi-C Handbook.
Data analysis
Hi-C data analysis is offered at our GeneGlobe Data Analysis Center. Hi-C sequencing results can be analyzed using the EpiTect Hi-C Data Analysis Portal, which uses open-source tools to provide a QC sequencing report, Hi-C contact matrices and visualization of chromatin contact maps. For more information see our EpiTect Hi-C Data Analysis Portal User Guide.
Chromatin conformation
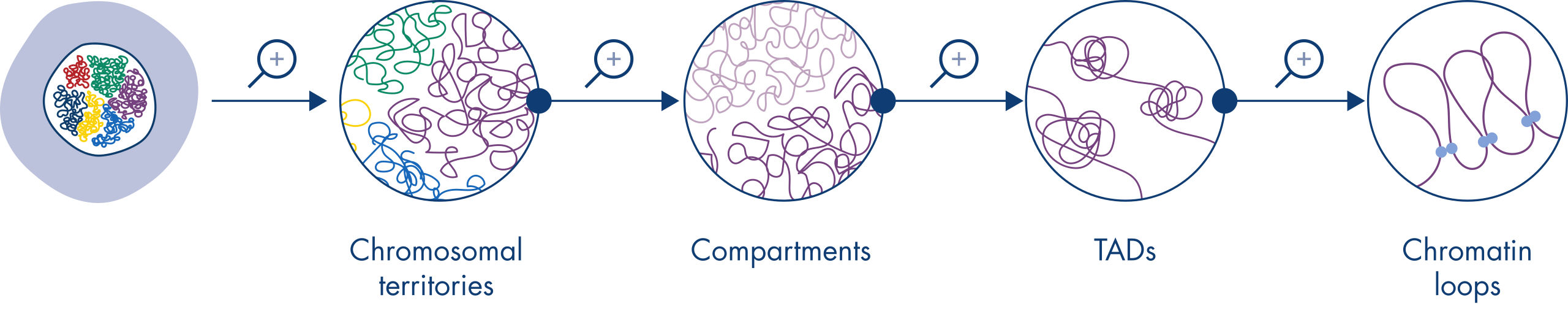
Hi-C has quickly become a very important tool for the analysis of nuclear organization. Analysis of Hi-C data has revealed the amazing complexity of genome architecture, with multiple layers of spatial organization that partition the genome into chromosome territories, chromosomal sub-compartments, topologically associated domains (TADs) and DNA loops at increasing resolution (see figure Levels of chromatin organization). In addition, genome organization is dynamic and changes during development. In no two cell types do chromosomes fold alike.
Chromosomal rearrangements and copy number variants
Individual chromosomes are physically separated into discrete territories and, therefore, DNA interactions captured by Hi-C primarily take place between DNA from the same chromosome (in cis) with little interaction between chromosomes (in trans). Owing to this phenomenon, Hi-C can be used as a genome-wide assay to identify translocations and other structural variants of interest. Compared to other NGS techniques, Hi-C requires extremely low coverage, which can save on costs. Furthermore, rearrangements involving poorly mappable regions can be better detected using Hi-C than standard NGS methods. Conveniently, the same Hi-C data can be used to detect copy number changes.
Genome assembly – haplotype phasing
When sequencing and assembling the genomes of new species, generating sequence scaffolds is often limited by large stretches of repetitive sequences that extend beyond the range of sequencing. In Hi-C data, the vast majority of interactions occur in cis between loci on the same chromosome. Additionally, a significant portion of these cis interactions is long-range, occurring between loci separated by millions of bases of DNA. These properties of chromatin interactions can be leveraged to order, orient and join sequence scaffolds into near full-length chromosomes without the need for a reference genome. Using the same principles, Hi-C interaction maps can be used to create diploid genomes by assigning genetic variants to paternal and maternal sister chromosomes (see figure Downstream applications of Hi-C sequencing data).